Définition des spécifications
Établir statistiquement les limites des paramètres design, procédé, matière et environnement — sur preuve, pas à dire d'expert.
Validation & qualification des procédés
De la caractérisation à la démonstration industrielle : établir les bonnes limites, prouver la performance lot après lot et industrialiser un procédé robuste — pire cas inclus.
Établir statistiquement les limites des paramètres design, procédé, matière et environnement — sur preuve, pas à dire d'expert.
Démontrer que le produit conserve ses performances dans le temps et résiste aux contraintes logistiques — dimensionnement des prélèvements et estimation du shelf life.
Identifier les paramètres critiques et leurs fenêtres opératoires, puis éprouver le procédé en conditions worst-case.
Démontrer la répétabilité lot après lot avec des plans d'échantillonnage dimensionnés sur le risque, pas sur la taille de lot.
Qualifier statistiquement les matières premières et construire une réception progressive — renforcée puis allégée.
* CQA — critical quality attribute · CPP — critical process parameters
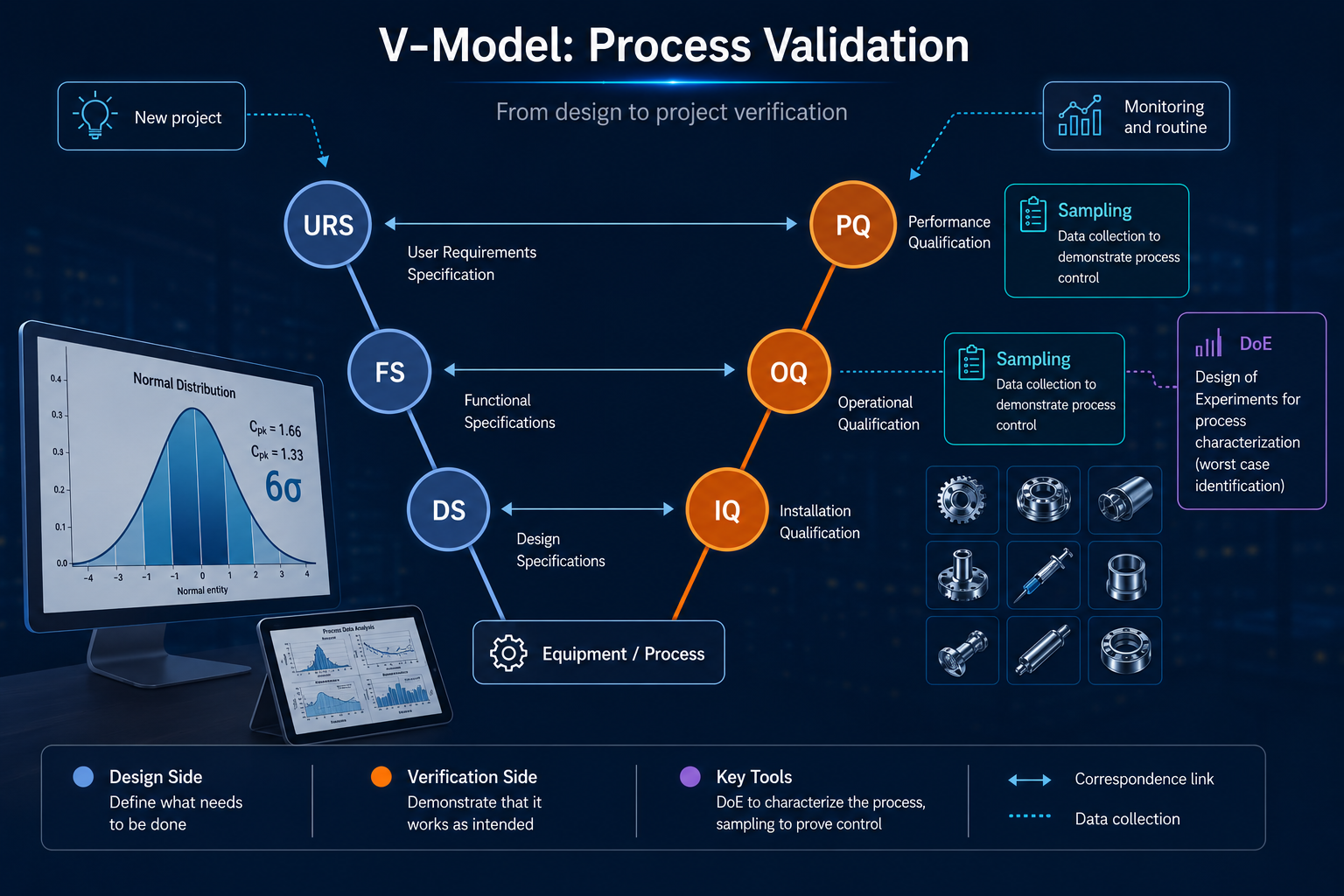
Validation de procédé : prouver que la conformité n'est pas une coïncidence.
Une validation de procédé n'est pas une formalité documentaire — c'est la démonstration, fondée sur le risque produit et procédé, que votre procédé produira systématiquement des unités conformes, y compris dans les conditions les plus défavorables. De la caractérisation jusqu'au plan de contrôle en routine, chaque étape — IQ, OQ, PQ — est dimensionnée sur le risque : taille de prélèvement et critère d'acceptation ne sont jamais arbitraires.
La même rigueur s'applique en amont, côté fournisseurs : qualifier statistiquement les matières premières et les composants, c'est s'assurer que la conformité du procédé ne repose pas sur des matières insuffisamment maîtrisées.
Enfin, démontrer que le produit tient dans le temps et résiste aux contraintes logistiques est une exigence à part entière — études de stabilité, vieillissement accéléré, simulation de transport — avec ses propres plans de prélèvement, critères d'acceptation et modélisation du shelf life.
C'est cette cohérence de bout en bout qui fait la différence entre un dossier qui passe un audit et un dossier qui le subit.
Un procédé validé ne se décrète pas — il se démontre. La question n'est pas « mon procédé a-t-il produit des pièces conformes lors des essais ? » mais « peut-on prouver, avec un niveau de confiance défini, qu'il produira systématiquement des pièces conformes — y compris dans les conditions les plus défavorables ? » C'est cette nuance qui fait la différence entre un dossier qui passe un audit et un dossier qui le subit.
Les spécifications ne tombent pas du ciel. Mal dimensionnées en amont, elles deviennent impossibles à tenir en production — ou tellement larges qu'elles ne protègent plus le patient. L'analyse statistique de tolérances et le robust design permettent d'établir des limites fondées sur la réalité du procédé et de la variabilité des matières, pas sur des jugements d'expert non documentés.
Avant même de qualifier le procédé, il faut démontrer que le produit conserve ses performances dans le temps et résiste aux contraintes logistiques. Ces études posent deux questions statistiques distinctes.
La première est celle du plan de prélèvement : combien d'unités inclure dans un protocole de stabilité ou de simulation de transport, et quel critère d'acceptation retenir, pour que les conclusions soient statistiquement défendables ? La taille de prélèvement et le critère d'acceptation sont définis par le risque produit et procédé — ce dimensionnement, trop souvent négligé, conditionne directement la validité des résultats face à un auditeur.
La seconde est celle de la modélisation du shelf life : à partir des données de vieillissement accéléré ou en temps réel, estimer la durée de vie du produit avec un niveau de confiance défini. Cette modélisation — qui s'appuie sur des techniques de régression et d'estimation statistique — permet de justifier la date de péremption sur des bases objectives, et non sur une simple extrapolation linéaire.
Avant l'OQ, l'étude de caractérisation — appuyée sur un plan d'expériences (DOE) — permet d'identifier les paramètres critiques du procédé et de cartographier leurs effets sur les réponses clés. C'est à ce stade que l'on détermine les fenêtres opératoires et que l'on identifie les conditions worst-case qui seront challengées lors de l'OQ. Sans cette étape, l'OQ revient à tester des conditions choisies arbitrairement — ce qui fragilise l'ensemble du dossier.
L'OQ confirme que le procédé reste conforme aux limites de ses paramètres opératoires, y compris dans les conditions les plus défavorables identifiées lors de la caractérisation. C'est une étape confirmatoire, pas exploratoire — elle s'appuie sur ce que la caractérisation a révélé.
La PQ est l'étape de démonstration industrielle. Elle apporte la preuve statistique que le procédé est reproductible à l'échelle de production réelle. La taille de prélèvement et le critère d'acceptation sont définis par le risque produit et procédé — pas par la taille de lot, ni par une convention arbitraire. C'est précisément ce lien explicite entre risque et plan statistique que les auditeurs FDA et les organismes notifiés européens vont chercher dans le dossier.
La PQ n'est pas une fin en soi — c'est le point de départ de la production courante. Une fois la validation acquise, il s'agit de mettre en place un plan de contrôle dimensionné au juste nécessaire : quelles caractéristiques produit et procédé surveiller, à quelle fréquence, avec quelle taille de prélèvement et quel critère d'acceptation. Là encore, c'est le risque produit et procédé qui dicte ces choix — en concentrant l'effort de contrôle sur les caractéristiques critiques identifiées lors de la caractérisation, sans surcharger la production.
La maîtrise d'un procédé commence par la maîtrise des matières qui l'alimentent. Le contrôle réception des matières premières et des composants s'appuie sur des référentiels reconnus — ISO 2859-1 pour les données attribut, ISO 3951-1 pour les données variables — qui permettent de justifier rigoureusement la taille de prélèvement et le critère d'acceptation en fonction du risque produit et procédé associé à chaque matière.
Mais l'objectif n'est pas de contrôler le maximum — c'est de contrôler le juste nécessaire. Deux leviers permettent d'y parvenir : la dynamisation, qui ajuste le niveau de contrôle en fonction de l'historique fournisseur, et le contrôle progressif, qui permet de minimiser la taille de prélèvement en cours de contrôle en fonction de la qualité observée sur le lot en cours — si les premiers résultats sont bons, on réduit ; si des non-conformités apparaissent, on renforce. Un fournisseur qui a prouvé sa régularité ne mérite pas le même niveau de surveillance qu'un fournisseur entrant — et le dossier doit pouvoir le justifier statistiquement.
Dimensionner chaque étape de votre validation — stabilité, caractérisation, OQ, PQ, plan de contrôle, réception fournisseurs — sur le risque produit et procédé et produire un dossier défendable de bout en bout, c'est l'accompagnement que propose StatExelia.
Parlons de votre projetDécrivez votre procédé et vos contraintes réglementaires. Nous proposons une stratégie de qualification proportionnée au risque.